MaDCaT – Mapping of Distances for the Categorization of Topology
Getting MaDCaT
MaDCaT is written in C++, with the source code freely available to academic users under the terms of the GNU General Public License. Download the source code from here. Inquiries about commercial licensing should be directed to Dr. Gevorg Grigoryan.
Downloading the distance-map database
MaDCaT uses the distance-map representation of structure to search. This means it needs a database of pre-computed maps to search against. Included with the source code is a program for generating maps from PDB structures. In addition, a database of maps derived from the PDB (circa 2012) can be downloaded from our server (includes only X-ray entries). To do so, issue the following command (this is a large file, ~71GB):
wget https://grigoryanlab.org/downloads/biounitMaps.tar.gz
To search against a database of maps, you will need to pass to MaDCaT a file with a list of paths to individual maps that you want it to consider (can be either full or relative to where you will be running MaDCaT from).Documentation
All the executables and scripts that are part of MaDCaT print detailed usage statements when run without command-line arguments. These contain the description of all allowed options. Below we present a simple usage scenario, which should cover the most typical circumstance. Some of the additional non-default options are pointed out along the way, but for a full list of all capabilities see the usage summaries printed by each program.
Suppose we have constructed an ideal fragment of tertiary structure, consistent of an alpha helix packing against a parallel two-stranded beta sheet:
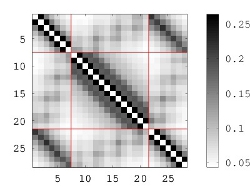
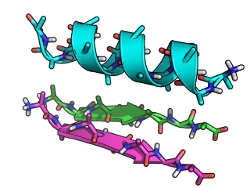 |
 |
$MADCAT_HOME/createDM --p motif.pdb --o motif.map
This generates a flat-text file motif.map which contains the distance matrix (in the first section of the file, before the END statement) as well as some additional information useful for MaDCaT. If you examine motif.map you will find that 1) values listed are inverse distances and 2) only atom pairs closer than 25 Å are listed (i.e. those with inverse distances below 0.04). Inverse distances below this cutoff value are assumed to be too small and are disregarded. 25 Å is the default cutoff and this can be adjusted using the --dcut option to createDM. Note, however, that the pre-generated database of PDB maps from our server has been constructed using the 25 Å cutoff, so this value should be used when searching against this database. The resulting distance map is visualized in the figure above on the right.The next step is to search a database with this map. Here the bc-30 database is used, which is highly non-redundant but still contains a large number of entries (~16,000 structures at the time of this writing):
$MADCAT_HOME/madcat --map motif.map --compList bc-30.list --diag 1 --topN 1000 --matchOut motif.match --seqOut motif.seq --structOut motif.struct --structOutType match
This tells MaDCaT to look into the file bc-30.list (assumed to be in the local directory here, but you can specify a path) for a list of database maps, search each for structural motifs similar to that given by motif.map, find the top 1,000 closest matches (in the sense of the distance-matrix difference norm) and produce several types of output. File motif.match (the "match" file) is the only mandatory output and it contains the exact specification of the 1,000 best matches found. This format is convenient for MaDCaT's internal use and this file can be used to reconstruct all of the other output information from a previous run. Here is a small fragment of the match file from the above run:
match: structure 'maps/a0/4a0f.1.bin' [(1: 906 x 906), (2: 906 x 769), (3: 906 x 787), (4: 769 x 906), (5: 769 x 769), (6: 769 x 787), (7: 787 x 906), (8: 787 x 769), (9: 787 x 787)] 0.0112288
match: structure 'maps/a0/4a0f.1.bin' [(1: 162 x 162), (2: 162 x 22), (3: 162 x 43), (4: 22 x 162), (5: 22 x 22), (6: 22 x 43), (7: 43 x 162), (8: 43 x 22), (9: 43 x 43)] 0.0112327
match: structure 'maps/l1/1l1s.1.bin' [(1: 1 x 1), (2: 1 x 14), (3: 1 x 32), (4: 14 x 1), (5: 14 x 14), (6: 14 x 32), (7: 32 x 1), (8: 32 x 14), (9: 32 x 32)] 0.0114376
...
0.810112 0.0112288 ASP LEU LEU CYS LEU VAL GLU LYS THR LEU VAL SER THR GLY ILE ALA ALA SER PHE LEU LEU THR LYS LEU LEU TYR LEU LYS
0.81016 0.0112327 ASP LEU LEU CYS LEU VAL GLU LYS THR LEU VAL SER THR GLY ILE ALA ALA SER PHE LEU LEU THR LYS LEU LEU TYR LEU LYS
0.797668 0.0114376 TYR ARG VAL VAL PHE HIS ILE ARG VAL LEU LEU LEU ILE SER ASN VAL ARG ASN LEU MSE ALA VAL ARG ILE GLU VAL VAL ALA
...
The last category of output are structures. In the syntax above, MaDCaT was asked to place the matching portion of structure from each match into a separate file under the directory named motif.struct (will be created if does not exist). Individual file names of matches are numbered to make the correspondence between match structures and lines in the above output files obvious. If the value of the option --structOutType includes the string "file", instead of one file per match, all matches will be put into a single NMR-style PDB file named according to the --structOut option.
Additional options exist that control whether and how information about sequences/structure intervening the disjoint query fragments in the matching structures is presented. See usage statements for more details.
By looking at the sequence output file, we can discern how close structures in real proteins come to our idealized query from above, as well as what sequences tend to stabilize them. As mentioned above, this file can (and often will) contain some redundancy (i.e. different matches may have the same sequence, usually indicating that they come from symmetric regions of a homo-oligomeric protein or a protein with an internal repeat). The perl script seqAnal.pl, included with MaDCaT, can be used to analyze these data by removing such redundancy. For example, the figure below shows the structures of all of the sequence-unique hits with an RMSD from the query below 0.8 Å:
|
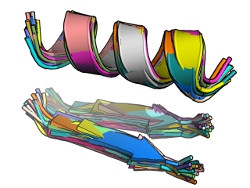 |
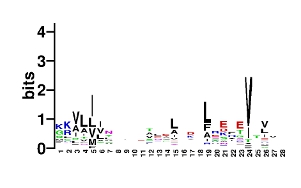 |
 |
perl -w $MADCAT_HOME/scripts/seqAnal.pl -s motif.seq -c 1.5 --sdir motif.structs --rbeg 1 --rlen 28 --rmsc 0.8 -o motif.eps
This asks seqAnal.pl to consider all unique matches to the query with an RMSD below 1.5 Å, then cluster those with a distance cutoff of 0.8 Å (greedy clustering is used), and output various information for the resulting clusters (the sequence logo, if WebLogo is installed, which matches make up the cluster, and their sequences) into file names based on the base name provided in the -o option. Switches --rbeg and --rlen designate the beginning and length of the region of structure used in clustering. The query structure is in our case 29 residues, so the entire structures is considered in clustering. But sometimes it is convenient to focus on variations only in a certain part of the structure. Many more options are available, please see the usage statement.A web-based search tool
A web-based applet for MaDCaT allows to search for a structural fragment, specified as a PDB file, with optional breaks in the structure. To limit server load, the web interface is currently limited to searching against a randomly sub-sampled set of non-redundant structures from the PDB (currently 1,000 structures) and returns the top 1,000 hits. Results from such a search can be used to determine whether a given structural motif is common, but will not find every matching instance of it in the PDB. For the latter, use of the stand-alone program is recommended.
Reference
If you use MaDCaT in your research, please cite the following paper:
Zhang J., Grigoryan G., "Mining Tertiary Structural Motifs for Assessment of Designability", Methods in Enzymology, 523: 21-40, 2013.