• Getting MASTER
• MASTER v.2 API
• Benchmarks
• Documentation
• Download database
• MASTER paper
Getting MASTER
MASTER is written in C++, with the source code freely available to academic users under the terms of the GNU General Public License. Click here to download MASTER as source code or binaries. Inquiries about commercial licensing should be directed to Dr. Gevorg Grigoryan.
MASTER v.2 API
MASTER v.2 features an entirely new and improved search algorithm that is available in the form of a C++ API library, thereby providing programmatic access to structure search functionality. See our paper by Zhou et al. [2] for more details. MASTER v.2 is freely available, in open-source form, for non-commercial purposes. Follow this link to request MASTER v.2.
Benchmark of Search Time
As detailed in Zhou and Grigoryan [1], MASTER search times were benchmarked on the following structural fragments of varying size, secondary-structural content, and number of disjoint segments:
Specifically, the fragments had between 6 and 50 residues and between 1 and 5 disjoint segments. The benchmark was performed on a machine with a 2.7-GHz Intel Xeon processor, using a diverse subset of the PDB with 12,661 structures as the database. Importantly, the machine had enough memory to fit the entire database, and the quoted times correspond to search times only (i.e., running times after the database has already been cached in memory). Thus, these times are most applicable to a scenario where a single machine is consistently being used for searching (and thus the database always remains cached in memory). Alternatively, the database can be placed on a RAM disk to enforce this.
All searches were performed under a range of RMSD cutoffs (0.4, 0.6, 0.8, 1.0, 1.5, and 2.0 Å) and benchmark times (in seconds) are organized according to the number of matches that result as well as the number of segments in the query:
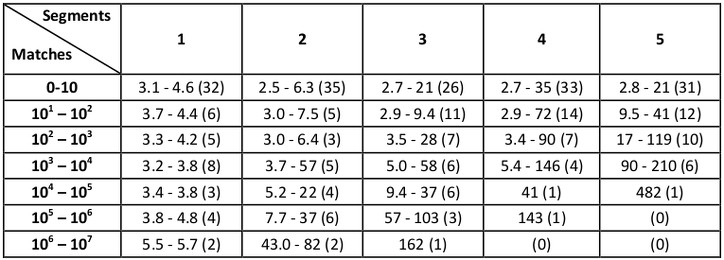
The numbers in parentheses indicate the number of examples in each category, and the shown ranges correspond to lowest and highest running times observed (in seconds). For example, for queries with two segments and RMSD cutoffs that resulted in 10 to 100 matches (there were five such examples in the benchmark set), running times ranged from 3.0 to 7.5 seconds.
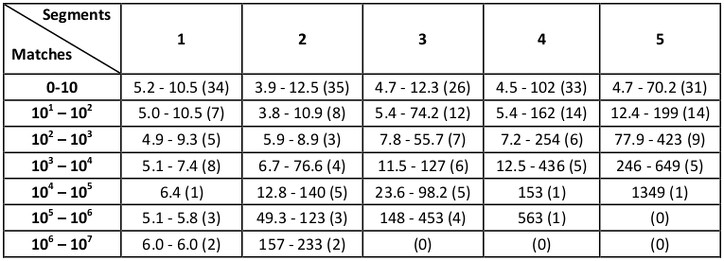
Starting with MASTER 1.3, we added the ability to search by full-backbone RMSD (i.e., RMSD calculated over N, C, CA, and O atoms) rather than just CA RMSD. Benchmark times for full-backbone RMSD searches (performed in the same way, with the same database, machine, and RMSD cutoffs as above) are:
As we describe in Zhou and Grigoryan [1], considerable speedups can be gained by using the --topN parameter in MASTER in addition to
the required RMSD cutoff. This allows one to specify the maximum number of top matches one could possibly be interested in. For example, if you know
you will not need to look at any more than 1000 closest matches, you can specify --topN 1000. In this way, as soon as MASTER finds 1000
matches that pass your RMSD cutoff, it can can safely begin making the cutoff more stringent while still guaranteeing to preserve the top 1000 hits.
For example, for both CA-only and full-backbone RMSD searches, specifying --topN 1000 results in speedups up to an order of magnitude in
cases where there would otherwise be many more matches.
The --topN parameter can also be a convenient "safety" feature. In that, if you do not have a good idea of what the right RMSD cutoff
for your search might be, and you thus risk over-estimating to produce millions (or more) matches, you can always go with a higher estimate of the
cutoff and specify a reasonable --topN, such that neither the number of matches nor the running time explode.
Documentation and Tutorials
Installation
MASTER is pretty easy to compile on Linux or MacOS. The source code distribution includes an INSALL file with compilation instructions. Also, the video here goes through this process in detail.
Building a Database
Once you have got MASTER compiled, the first step to using it is to create a database you will be searching. See this video for a detailed description of this process.
Usage Example 1
In this example we analyze a motif taken from a protein-peptide interface. We first define and search the motif and then analyze the results.
Usage Example 2
Coming soon...
Download a MASTER database
If you do not want to build your own database (although it is advisable to do so because the "right" database to use will depend on your application), you can download our database to get going quickly. This database is made up of non-redundant chains produced by clustering all chains in the PDB with blastclust and taking he representative chain from each cluster. This database was built using the PDB as of 10/22/2014. Note that this is a single-chain database. This means that even PDB entries that have multiple chains in their biological unit will be split into separate entries containing the individual chains. This may or may not be ok for your applications. To download the database use the following command:
wget https://grigoryanlab.org/downloads/masterDB.tar.gz
To search against this database, specify the list file list (which you will find inside the directory once you uncompressed the tar archive) as the parameter --targetList in MASTER. NOTE: you will need to edit this file to replace the base path with where you store your maps locally.Reference
If you use MASTER in your research, please cite the following paper:
[1] Zhou J., Grigoryan G., "Rapid Search for Tertiary Fragments Reveals Protein Sequence-Structure Relationships", Protein Science, 24(4): 508-524, 2015.
[2] Zhou J., Grigoryan G., "A C++ library for protein sub-structure search", Bioinformatics (under review)